Protein from the food we eat is broken down into building blocks of protein (amino acids) which are then further metabolised by specific enzymes for body growth and energy production.
3-hydroxy-3-methylglutaryl-CoA lyase deficiency (also known as HMG-CoA lyase deficiency) is an inherited organic acid disorder in which the body is unable to process an amino acid called "leucine" due to the deficiency of an enzyme called "HMG-CoA lyase". This enzyme is responsible for breaking down dietary proteins and fats for energy. It also produces ketone during the breakdown of fats. Ketones are the important sources of energy for our body, especially the brain, during periods without food (fasting).
In this condition when the body cannot process leucine normally, buildup of organic acid can cause the blood to become too acidic (metabolic acidosis). In addition, a shortage of ketone leads to low sugar level (hypoglycaemia). Both metabolic acidosis and low blood sugar can result in cell damage, particularly in the brain.
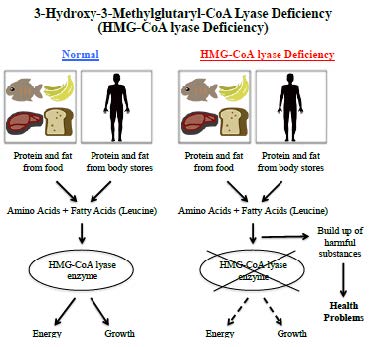
The original graphic is converted into the following text version for your easy access to the information.
3-hydroxy-3-methylglutaryl-CoA lyase deficiency (HMG- CoA lyase deficiency)
Our body breaks down protein and fats from food into amino acids and fatty acids when we eat, and breaks down protein and fats from body stores into amino acids and fatty acids during prolonged fasting and stress. When functioning normally, our body will produce HMG-CoA lyase to metabolise leucine and fatty acids into ketone to produce energy for our body's use and growth. Ketones are the important sources of energy for our body, especially the brain, during periods without food. In people with 3-Hydroxy-3-Methylglutaryl-CoA Lyase Deficiency, the HMG-CoA lyase is either missing or not working properly, so amino acids and fatty acids cannot be metabolized properly. As a result, harmful substances will build up in the body and cause serious health problems.
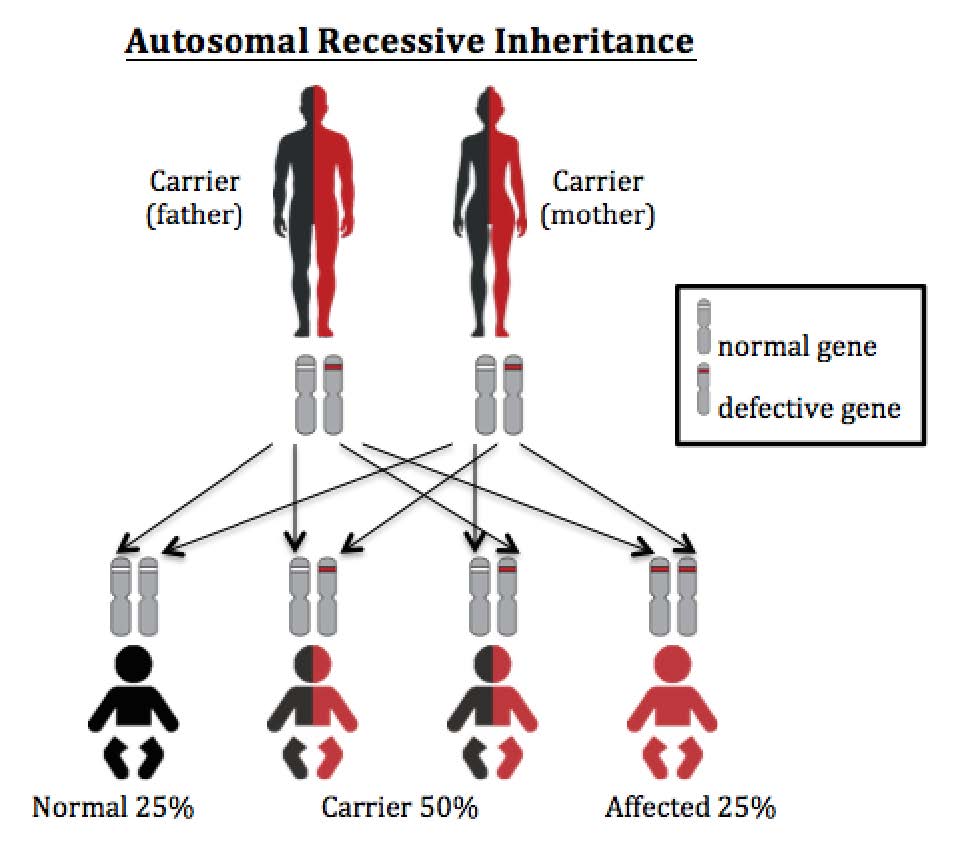
Everybody has two copies of genes, one from each parent, which tell the body how to make specific enzymes.
HMG-CoA lyase deficiency is an autosomal recessive disease. Only when babies inherit two faulty copies of the gene for HMG-CoA lyase deficiency from parents, the enzyme made does not work properly or is not even made at all.
The original graphic is converted in to the following text version for your easy access to the information.
Autosomal recessive inheritance
In autosomal recessive diseases, people with two faulty copies of gene (one from father and one from mother) will develop symptoms. People with only one faulty copy of gene are normal and they are called disease carriers. HMG- CoA lyase deficiency is inherited in autosomal recessive manner.
If both parents are HMG- CoA lyase deficiency carriers, for each pregnancy (no matter it is a baby boy or girl), there is a 25% (1 in 4) chance that the child has 2 copies of normal gene (who is not affected), a 50% (1 in 2) chance that the child has one normal and one faulty gene who is a carrier like the parents, and a 25% (1 in 4) chance that the child has two copies of faulty gene who is at risk for HMG- CoA lyase deficiency.
Babies with HMG-CoA lyase deficiency may experience slightly different signs and symptoms. Most affected children start to show signs at 3 months to 2 years of age though few may show signs of the condition just a few days after birth. Metabolic crisis, which is a period of time when a metabolic disorder makes the baby seriously ill, may develop and such episode is frequently triggered by infection, prolonged period without food or increased intake of protein-rich food.
With early and careful treatment, babies with HMG-CoA lyase deficiency can have healthy growth and development. It is possible, even with treatment, for babies with HMG-CoA lyase deficiency to have low blood sugar and other signs of HMG-CoA lyase deficiency.
Early screening and treatment of HMG-CoA lyase deficiency are very important and babies who do not receive treatment usually die or develop permanent brain damage.
The treatment for HMG-CoA lyase deficiency may include:
It is important that babies with HMG-CoA lyase deficiency need to be fed regularly and should not go for long periods without eating (i.e. fasting). They also need to see their metabolic doctors regularly.
If you are worried that your baby is ill, it is important to follow medical advice. Bring your baby to your local accident and emergency department immediately. Take any information that you have been given about HMG-CoA lyase deficiency, including this pamphlet, to the hospital with you.
For general queries on Newborn Screening Programme for Inborn Errors of Metabolism, please call 5741 4280 (Department of Clinical Genetics, Hospital Authority)
July 2024
Hospital Authority