PKU is a rare but treatable amino acid disorder. People with amino acid disorders cannot process amino acids, the building blocks of protein.
Our body breaks down protein in food into amino acids when we eat and breaks down protein in our muscle into amino acids during prolonged fasting and stress.
Amino acids are then processed by special chemicals called enzymes so that the body can use them. Different enzymes target specifically at different amino acids.
Babies with PKU lack the specific enzyme called "phenylalanine hydroxylase" (PAH) which is responsible to process an amino acid called phenylalanine into another amino acid called tyrosine.
When phenylalanine hydroxylase is missing or malfunctioning, phenylalanine accumulates to a harmful level. Tyrosine, which is essential for proper brain functioning, may be insufficient. This causes long term health problems including learning difficulties.
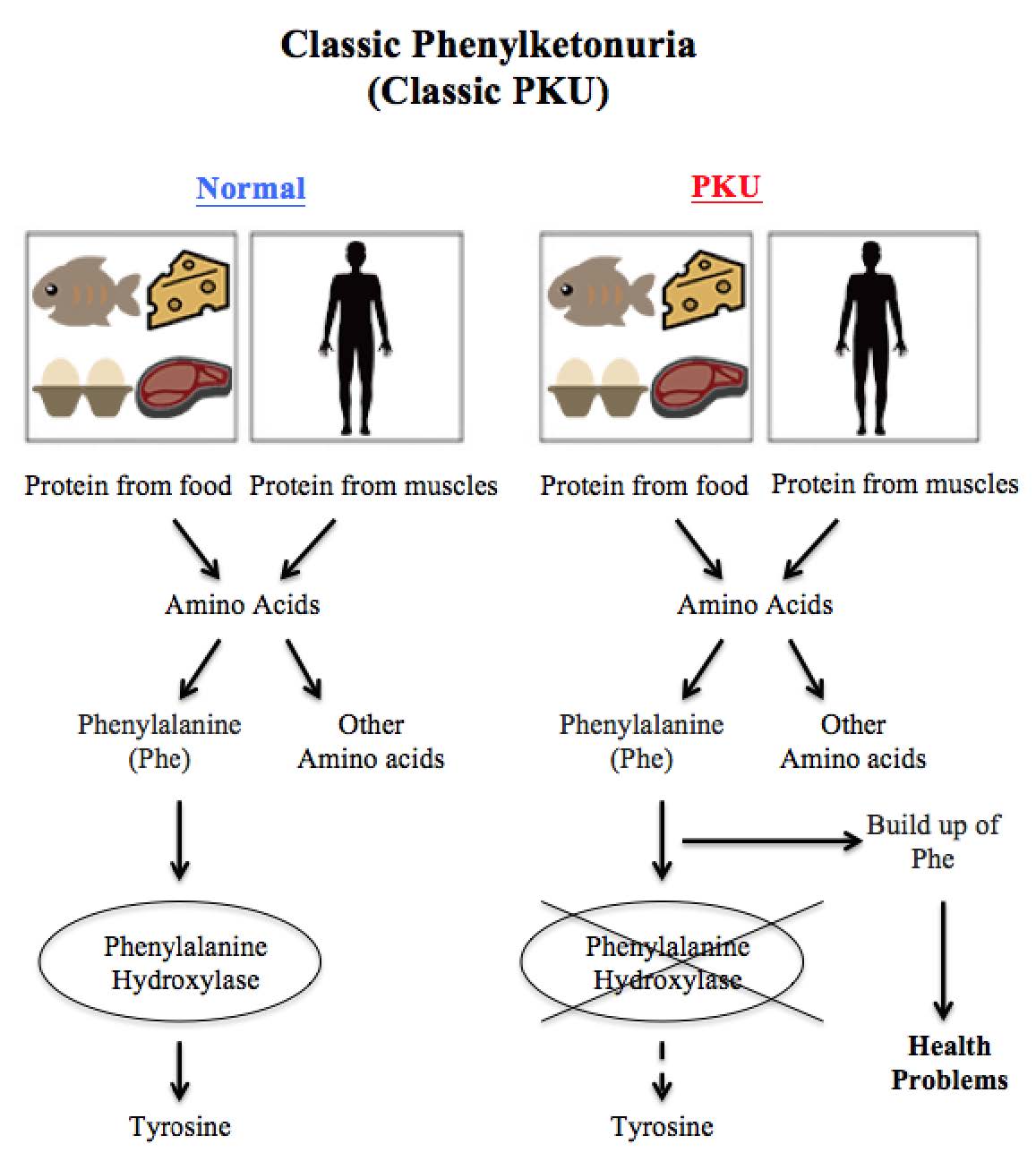
The original graphic is converted into the following text version for your easy access to the information.
Classic Phenylketonuria (Classic PKU)
Our body breaks down protein in food into amino acids when we eat and breaks down protein in our muscles into amino acids during prolonged fasting and stress. When functioning normally, our body will produce phenylalanine hydroxylase (PAH) to convert amino acid phenylalanine into the amino acid tyrosine, so that amino acids can be utilized properly for our body's use and growth. In people with PKU, the PAH enzyme is either missing or not working properly, making the amino acid phenylalanine cannot be metabolized properly. As a result, phenylalanine will build up in our body to a harmful level and cause serious health problems.
Everybody has two copies of genes, one from each parent, which tell the body how to make specific enzymes.
PKU is an autosomal recessive disease. Only when babies inherit two faulty copies of the gene for PKU from parents, the enzyme made does not work properly or is not even made at all.
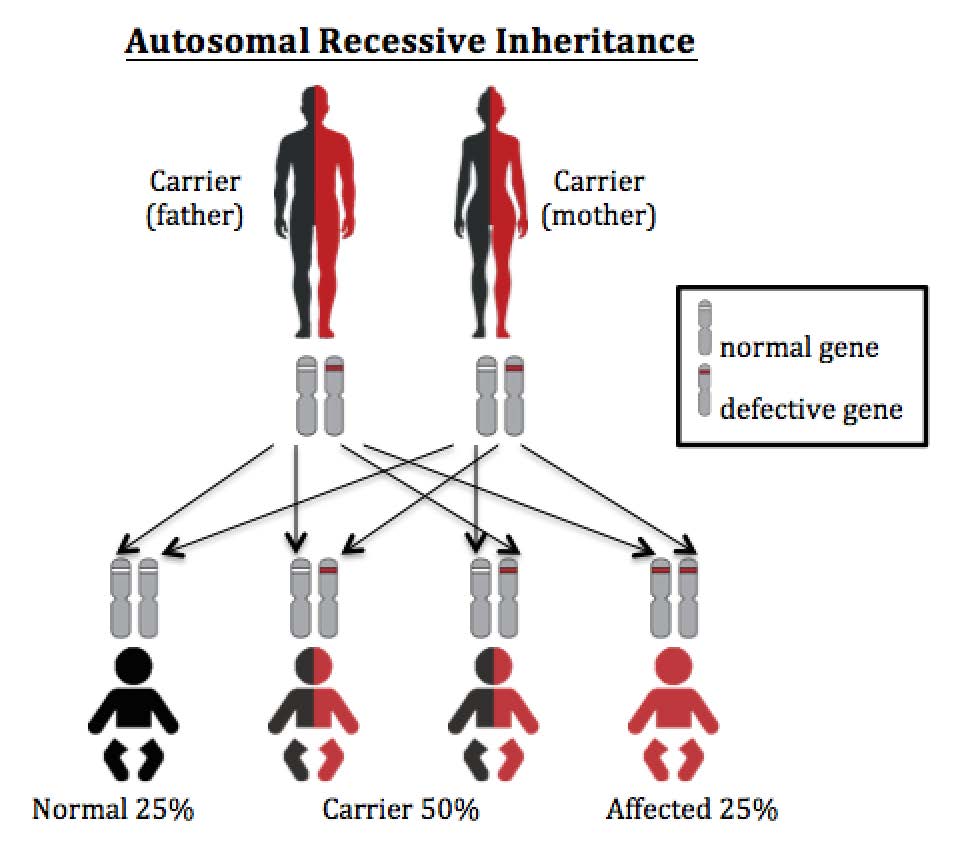
The original graphic is converted in to the following text version for your easy access to the information.
Autosomal recessive inheritance
In autosomal recessive diseases, people with two faulty copies of gene (one from father and one from mother) will develop symptoms. People with only one faulty copy of gene are normal and they are called disease carriers.
PKU is inherited in autosomal recessive manner. If both parents are PKU carriers, for each pregnancy (no matter it is a baby boy or girl), there is a 25% (1 in 4) chance that the child has 2 copies of normal gene (who is not affected), a 50% (1 in 2) chance that the child has one normal and one faulty gene who is a carrier like the parents, and a 25% (1 in 4) chance that the child has two copies of faulty gene who is at risk for PKU.
Babies with PKU are usually healthy at birth. They gradually develop symptoms during infancy as a result of the accumulation of phenylalanine. This includes progressive and irreversible brain damage, leading to neurological disorders, learning difficulties and behavioural problems.
If right treatment is started early, babies with PKU are well and can have healthy and active lives.
Uncommonly, some babies have the condition called "mild hyperphenylalaninaemia", with only slightly increased phenylalanine levels in the blood. They do not have classic PKU. They might not need any treatment and only need regular monitoring of phenylalanine levels in the blood.
PKU can be treated with special low phenylalanine diet and supplements which reduce the build-up of phenylalanine in the body and prevent irreversible brain damage. Dietary treatment should start as early as possible and continue throughout the life.
Babies with PKU need to take a special nutritionally balanced phenylalanine-free infant formula. High protein foods including milk (both breast milk and normal infant formula) have to be limited in order to provide small amount of phenylalanine just right for normal growth, development and health.
Patients with PKU need to see their specialist metabolic team regularly even when they do not have the symptoms. They need to have regular monitoring of phenylalanine levels in the blood. The metabolic team will adjust the diet accordingly.
If you are worried that your baby is ill, it is important to follow medical advice. Bring your baby to your local accident and emergency department immediately. When you are going to the hospital, take the special infant formula and any information that you have been given about PKU, including this pamphlet, with you.
For general queries on Newborn Screening Programme for Inborn Errors of Metabolism, please call 5741 4280 (Department of Clinical Genetics, Hospital Authority)
July 2024
Hospital Authority