精氨基琥珀酸血症是一种罕见但可治疗的氨基酸代谢障碍。患者不能适当地处理构成蛋白质的基本单元 — 氨基酸。
当我们进食时,身体将食物中的蛋白质分解成氨基酸;在过饥或应激状态下,我们肌肉中的蛋白质亦会被分解成氨基酸。过量的氨基酸会转化成对脑部极之有害的阿摩尼亚。身体有一套叫「尿素循环」的机制,将阿摩尼亚快速转化成尿素,然后随尿液排出体外。这个由六种特别的酶所组成的排毒机制,亦负责产生一种名为「精氨酸」的氨基酸。
患上精氨基琥珀酸血症的婴儿缺乏了尿素循环中的一种酶,名为「精氨基琥珀酸裂解酶」,因而令尿素循环不能有效地运作。结果,有害的阿摩尼亚积聚于体内,而维持血管健康的精氨酸含量亦变得不够。这导致长远的健康问题包括脑部损伤、高血压及肝脏疾病。
上述内容原以图表形式表达,为方便使用者获取有关资讯,已转换成纯文字版本,详情如下:
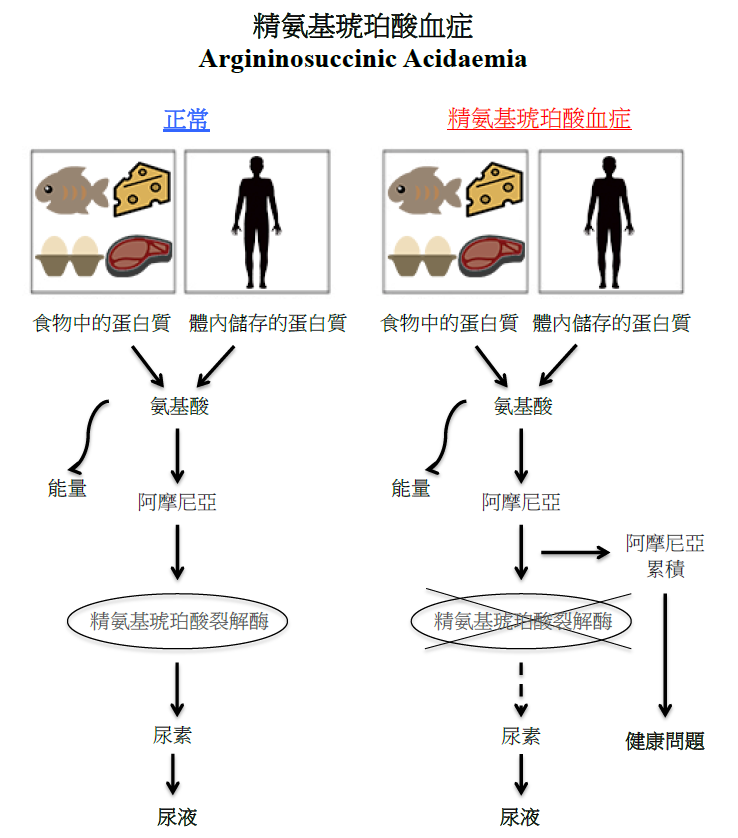
精氨基琥珀酸血症
当我们进食时,食物中的蛋白质会被分解成氨基酸,而在过饥或应激状态下,肌肉中的蛋白质亦会被分解成氨基酸。过量的氨基酸会转化成有害的阿摩尼亚。在正常情况下,身体会制造精氨基琥珀酸裂解酶,去帮助将有害的阿摩尼亚转化为尿素,以便随尿液排出体外。但在精氨基琥珀酸血症患者中,由于精氨基琥珀酸裂解酶的缺失或功能不正常,因此未能有效地将阿摩尼亚分解代谢,令有害的阿摩尼亚在体内积聚,引致严重的健康问题。
每个人都有一对基因,由父母各自遗传一条,基因指导身体制造所需的酶。
精氨基琥珀酸血症是经常染色体隐性遗传的疾病。只有当婴儿从父及母同时遗传两个精氨基琥珀酸血症的异常基因时,其身体所制造的酶便不能发挥正常功能,甚至乎身体完全不能制造所需的酶。
上述内容原以图表形式表达,为方便使用者获取有关资讯,已转换成纯文字版本,详情如下:
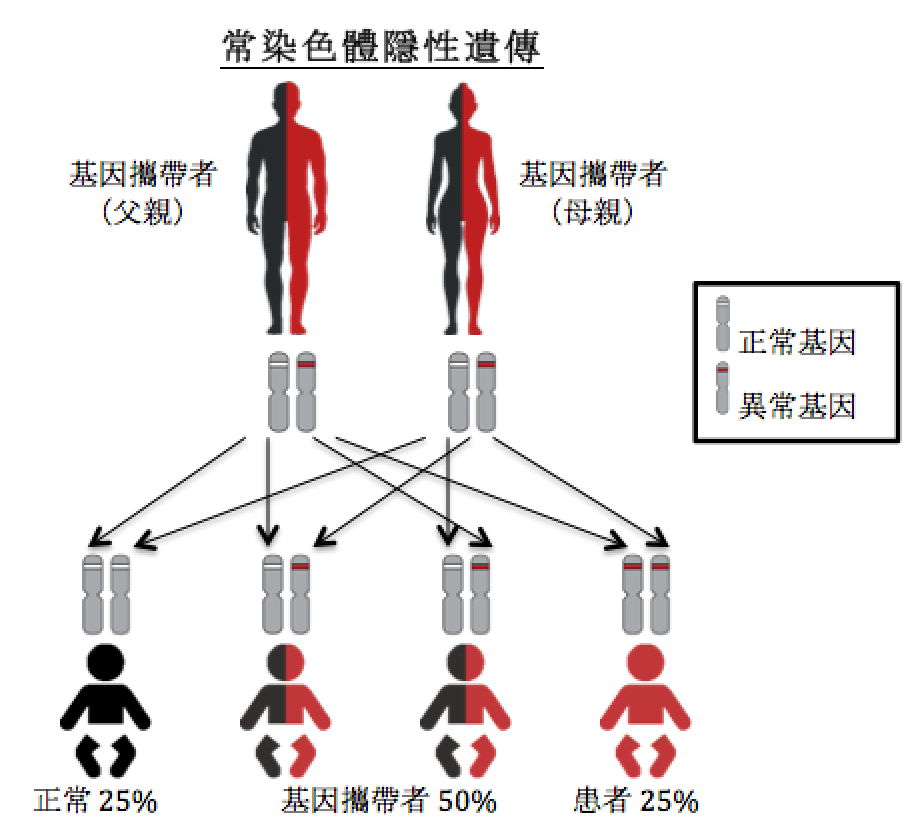
常染色体隐性遗传
就常染色体隐性遗传病,当患者遗传了两个病变基因(一个来自父亲,一个来自母亲)才会发病。如果一对基因中只有一个病变基因,他们则被称为基因携带者,不会有任何病征。精氨基琥珀酸血症是常染色体隐性遗传病。
假如父母都是精氨基琥珀酸血症的基因携带者,他们每次怀孕的孩子(不分男女)会有25%(即四分之一)的机会遗传了两个正常的基因,即没有精氨基琥珀酸血症;50%(即二分之一)的机会遗传了一个正常基因和一个病变基因,与父母一样,属于基因携带者;25%(即四分之一)的机会遗传了两个病变基因,成为精氨基琥珀酸血症患者。
患有精氨基琥珀酸血症的婴儿在出生时看似健康,但很快就会出现症状;因为阿摩尼亚可于出生后几天内大量积累,而导致急性代谢危机。
急性代谢危机是指患者因为代谢障碍,而导致病情在短时间内变得危重。当婴儿长时间没有进食或当出现感染、发烧或胃部不适时,会容易诱发急性代谢危机,如果没有及时接受治疗,患者可能会恶化至抽搐、昏迷,甚至危及生命。
如能及早得到适当治疗,患有精氨基琥珀酸血症的婴儿是可以过著健康的生活。
征状因人而异。有些患者病情轻微,甚至到成年还没有征状,但是出现急性代谢危机的机会仍然存在。有些患者从没有岀现急性代谢危机,但也有机会出现健康问题,例如肝脏纤维化、高血压、学习障碍或脑痫症等。
患有精氨基琥珀酸血症的婴儿须要接受低蛋白饮食治疗及服用精氨酸补充剂。重要的是要进食定时,避免过长时间禁食。有些病人同时需要药物治疗。
患有精氨基琥珀酸血症的婴儿于没有任何征状期间亦需往代谢科儿科医生进行定期覆诊。重要的是要预先编制一个特别的照顾方案,以便于生病期间或食欲不振时采用,以预防急性代谢危机。
如果你担心你的婴儿生病了,请谨记要按照医生的建议,立即将婴儿送到附近的急症室,以及将您所获得有关精氨基琥珀酸血症的药物、特别配方奶粉和任何资料(包括本小册子)随身携带到医院。
关于初生婴儿代谢病筛查计划的一般查询,可致电:
5741 4280 (医院管理局医学遗传科)
二零二四年七月版
医院管理局