卡尼丁结合酵素 II 缺乏症是一种遗传性脂肪酸氧化障碍。脂肪酸氧化障碍患者不能分解脂肪成为脂肪酸,从而制造能量。
脂肪酸是心脏和肌肉的主要能量来源。在禁食期间,脂肪酸也成为肝脏和其他器官的重要能量来源。脂肪酸要靠名为酶的特别化学物质去分解成为能量,不同的酶会分解特定不同的脂肪酸。当中「卡尼丁结合酵素第二型」(简称CPT-II) 这种酶主要负责处理分解「长链脂肪酸」。
当CPT-II不足或不能有效运作时,长链脂肪酸便不能被分解并转化为能量。当脂肪不能提供能量时,糖份便成为主要的能量来源。当糖份大量消耗时,低血糖症便会出现。此外,当身体不能有效地分解脂肪酸时,有害的代谢物会积聚于身体,造成健康问题。
上述内容原以图表形式表达,为方便使用者获取有关资讯,已转换成纯文字版本,详情如下:
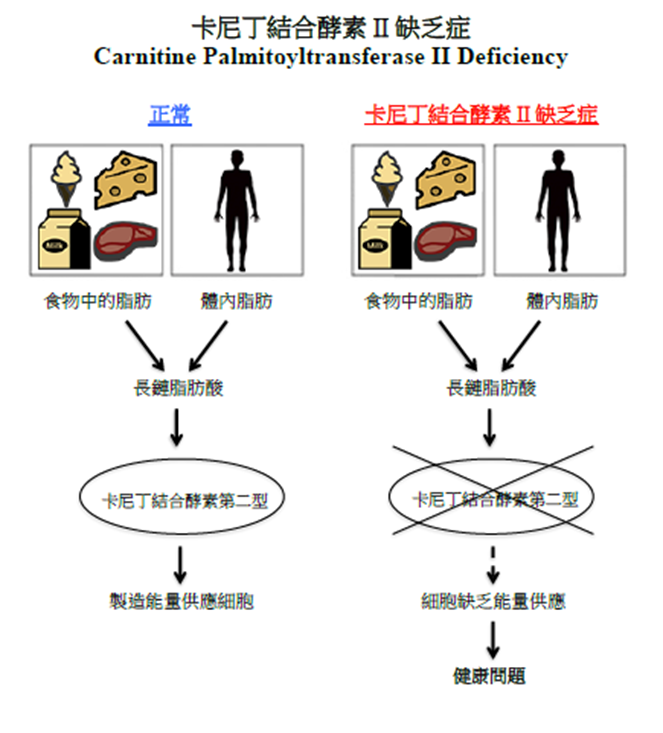
卡尼丁结合酵素 II 缺乏症
当我们进食时,食物中的脂肪会被分解成脂肪酸;而当过饥或在应激状态下,体内储存的脂肪亦会被分解成脂肪酸。在正常情况下,身体会制造一种名为「卡尼丁结合酵素第二型」的酶,以分解长链脂肪酸,去制造能量供应细胞所需。但在卡尼丁结合酵素 II 缺乏症患者中,由于卡尼丁结合酵素第二型的缺失或功能不正常,导致长链脂肪酸无法正常地被身体分解代谢为能量。结果使细胞缺乏能量供应,引致严重的健康问题。
每个人都有一对基因,由父母各自遗传一条。基因指导身体制造所需的酶。
卡尼丁结合酵素 II 缺乏症是经常染色体隐性遗传的疾病。只有当婴儿从父及母同时遗传两个卡尼丁结合酵素 II 缺乏症的异常基因时,所制造的酶便不能发挥正常功能,甚至乎身体完全不能制造所需的酶。
上述内容原以图表形式表达,为方便使用者获取有关资讯,已转换成纯文字版本,详情如下:
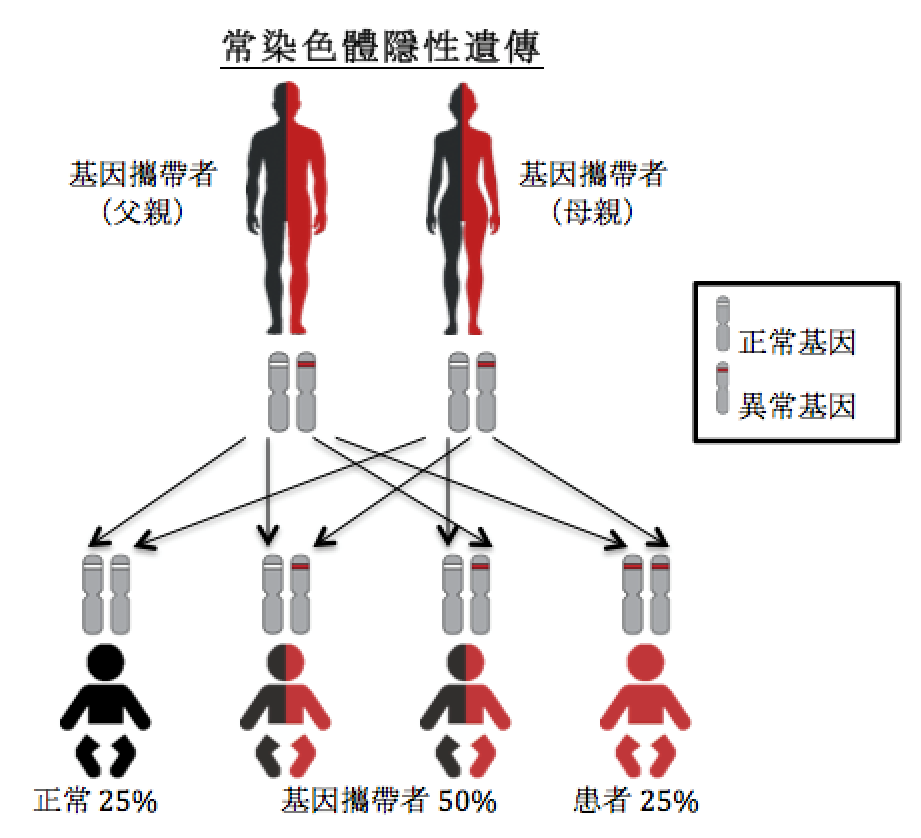
常染色体隐性遗传
就常染色体隐性遗传病,当患者遗传了两个病变基因(一个来自父亲,一个来自母亲)才会发病。如果一对基因中只有一个病变基因,他们则被称为基因携带者,不会有任何病征。卡尼丁结合酵素 II 缺乏症是常染色体隐性遗传病。
假如父母都是卡尼丁结合酵素 II 缺乏症的基因携带者。他们每次怀孕的孩子(不分男女)会有25%(即四分之一)的机会遗传了两个正常的基因,即没有卡尼丁结合酵素 II 缺乏症;50%(即二分之一)的机会遗传了一个正常基因和一个病变基因,与父母一样,属于基因携带者;25%(即四分之一)的机会遗传了两个病变基因,成为卡尼丁结合酵素 II 缺乏症患者。
大部份卡尼丁结合酵素 II 缺乏症的患者直至青少年期或成年初期都没有征状。只有部份重症患者在出生不久或婴儿期出现征状,甚至会发生急性代谢危机。
急性代谢危机是指患者因为代谢障碍,而导致病情在短时间内变得危重。感染、发烧、胃部不适或长时间没有进食,都会容易诱发急性代谢危机。
卡尼丁结合酵素 II 缺乏症的患者在青春期或成年初期常常出现肌肉乏力或痉挛,尤其在剧烈运动后、受泠后、禁食或受感染后。严重时,患者会因肌肉受损溶解引致尿液变红和肾衰竭。
更严重的患者也有可能出现以下的健康问题:
代谢病儿科医生及营养师会提供专家意见及适切治疗。治疗目的是维持足够能量供应身体,及避免有害的代谢物积聚而引发急性代谢危机。
重要的是,家长应定时向患有卡尼丁结合酵素 II 缺乏症的婴儿提供喂食,不能让他们长时间禁食。
事先与你的医生和营养师讨论和编制一套护理计划也是非常重要。这计划让你知道于生病期间或胃口欠佳时如何照顾你的婴儿,并提供额外的含糖食物,好好地预防急性代谢危机。
有需要时,患者可能要接受终生治疗:
如果你担心你的婴儿生病了,请谨记要按照医生的建议,立即将婴儿送到附近的急症室,以及将您所获得有关卡尼丁结合酵素 II 缺乏症的药物、特别配方奶粉和任何资料(包括本小册子)随身携带到医院。
关于初生婴儿代谢病筛查计划的一般查询,可致电:
5741 4280 (医院管理局医学遗传科)
二零二四年七月版
医院管理局